

- Learning process on protein sequences
- Data for the article Local Substitutability for Sequence Generalization (2012)
- Data for the article A bottom-up efficient algorithm learning substitutable languages from positive examples (2014)
Learning Process on Protein Sequences
We detail the experimentation process using on protein sequences.
The document here provides an overview of the automated pre-processing and post-processing steps necessary to apply the main generalization algorithm ReGLiS on protein sequences. When dealing with protein sequences, it is important to take into account the similarities between the 20 amino-acids (the alphabet \(\Sigma_{a} \) of proteins) arising from shared physico-chemical properties: some amino-acid replacements have no impact while others have an effect on the function or the structure of the protein. To take into account this knowledge directly in the learning sequences, a standard approach consists in recoding the proteins on a smaller property-based alphabet, such as the hydropathy index or the Dayhoff encoding. Instead of making use of these static coding schemes, this work has adopted a more specific data-driven approach. It relies on the detection of local similarities in the training set by building a partial local multiple alignment (PLMA) of the sequences. Each short strongly conserved region in the PLMA (also called block) will form one of the characters for recoding sequences.The computation of PLMA is also the first step performed in Protomata-Learner (Kerbellec(2008)). But whereas the choice of the alignment parameters is important in Protomata-Learner to tune the desired level of generalization, we have used only a basic set of parameters in this study (In practice, we have used the same following command line for all PLMA: paloma -i foo.fasta -o foo.plma -block-mode maxWeightFragments -transClosure -t 5 -M 7 -q 2 ).
Apart from blocks, it is necessary to take into account the existence of subsequences that appear in no block and this explains the rather complicated procedure to get a practical grammar directly able to parse protein sequences from the learned grammar. The reader not interested by these practical considerations on protein sequence parsing can safely ignore the following paragraph detailing the procedure steps.
PLMA are generated (step \(a \)) using Protomata-Learner, version 2.0(available at http://tools.genouest.org/tools/protomata/, which works on a set of protein sequences \(S \). PLMA form a set of, non-overlapping and non-crossing (i.e. consistent in the sense of Morgenstern(2001), alignment snippets on \(S \) named PLMA blocks. Each PLMA block is itself made up of a set of conserved substrings of same length from a subset of the training sequences (with only one substring per sequence involved in the block). A block implicitly aligns its substrings by their relative position, i.e. each position in a block corresponds to aligned characters for all of its sequences.
Sequences are recoded according to the PLMA blocks that are crossed by the sequence (step \(b.1 \)) and in parallel the information of the amino-acids composition of each block in retained in a grammar \(G_{a} \) (step \(b.2 \)). This is achieved by adding for each block \(B \) of length \(l \), a rule:
\(B\rightarrow P_{1}…P_{l} \),
and for each amino-acid \(A \) at position \(p \) in the block, a rule :
\(P_{p}\rightarrow N_{A} \).
It is in fact a little more complex since some a priori knowledge on proteins can be introduced in this grammar. Specifically, we added a simple error model on allowed sequences, based on the standard amino acid substitution matrix Blosum62, which scores the degree to which a given amino-acid can be substituted by another one without functional loss. Thus for each pair of amino-acid pair \((A,C) \) for which the score reflects a mutation that is more frequent than just by chance, the following rule is added:
\(N_{A}\rightarrow C \).
This is a neat way to artificially enlarge the size of the training sample that is usually too small to provide this fine-level variation information on protein sequences
The rationale behind block recoding is that non conserved substrings are likely to be uninteresting for the characterization of the protein family. Then, like in Protomata, one has to fix the treatment of substrings that belong to a sequence but not to a block. If a substring occurs between two blocks and no block is found between these two blocks in other sequences, it is considered as a gap and a rule is dedicated to its recognition:
\(Gap\rightarrow\Sigma_{a}Gap\vert\lambda \).
Other substrings that perform a shortcut of one or several blocks are called “exceptions”. In order to avoid overgeneralization, we have kept each exception \($ E_{0}\ldots E_{n} \) as a new block and added a dedicated rule in the grammar:
\(E\rightarrow N_{E_{0}}…N_{E_{n}} \).
Before applying ReGLiS on the recoded sequences, gaps are removed (step \(c.1 \)) to avoid a meaningless generalization (a context with only gaps is not significant). The immediate context (left and right block) of each gap is stored (step \(c.2 \)) for further reinsertion.
The main step \(d \) applies the algorithm on the PLMA block sequences without gaps and produces a core grammar \(G_{b} \).
Gaps are then reinserted in the grammar during step \(e \) by replacing each rule \(L\rightarrow…\alpha_{i}\alpha_{i+1}… \) in \(G_{b} \) where the last character of \(\alpha_{i} \) and the first character of \(\alpha_{i+1} \) form the context of a gap, by the rule:
\(L\rightarrow…\alpha_{i} \) \(Gap \)
\(\alpha_{i+1}… \)
This leads to a grammar \(G^{gap}_{b} \) that is then merged to \(G_{a} \) by changing terminals blocks into non terminals (step \(f \)).
-
Local Substitutability for Sequence Generalization, François Coste, Gaëlle Garet and Jacques Nicolas, ICGI 2012, Washington (United States)
paper, slides
Protein Family : PS00307
Cross-validation files
| split | training sequences | plma file | plma svg | test sequences |
|---|---|---|---|---|
| 0 | train0 | plma0 | plmasvg0 | test0 |
| 1 | train1 | plma1 | plmasvg1 | test1 |
| 2 | train2 | plma2 | plmasvg2 | test2 |
| 3 | train3 | plma3 | plmasvg3 | test3 |
| 4 | train4 | plma4 | plmasvg4 | test4 |
| 5 | train5 | plma5 | plmasvg5 | test5 |
| 6 | train6 | plma6 | plmasvg6 | test6 |
| 7 | train7 | plma7 | plmasvg7 | test7 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Grammar files obtained with different criteria
| split \ k,l | 1,1 | 2,2 | 3,3 | 4,4 | 5,5 | 7,7 | 10,10 | 15,15 | 20,20 | 25,25 | 30,30 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | |
| 0 | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 1 | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 2 | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 3 | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 4 | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 5 | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 6 | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 7 | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
-
A bottom-up efficient algorithm learning substitutable languages from positive examples, François Coste, Gaëlle Garet and Jacques Nicolas, ICGI 2014, Kyoto (Japan)
Protein Family : PS00219
Cross-validation files
| split | training sequences | plma file | plma svg | test sequences |
|---|---|---|---|---|
| 0 | train0 | plma0 | plmasvg0 | test0 |
| 1 | train1 | plma1 | plmasvg1 | test1 |
| 2 | train2 | plma2 | plmasvg2 | test2 |
| 3 | train3 | plma3 | plmasvg3 | test3 |
| 4 | train4 | plma4 | plmasvg4 | test4 |
| 5 | train5 | plma5 | plmasvg5 | test5 |
| 6 | train6 | plma6 | plmasvg6 | test6 |
| 7 | train7 | plma7 | plmasvg7 | test7 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Grammar files obtained with different criteria
| split \ k,l | 1,1 | 2,2 | 3,3 | 4,4 | 5,5 | 6,6 | 7,7 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | |
| 0 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 1 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 2 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 3 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 4 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 5 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 6 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 7 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
Protein Family : PS00063
Cross-validation files
| split | training sequences | plma file | plma svg | test sequences |
|---|---|---|---|---|
| 0 | train0 | plma0 | plmasvg0 | test0 |
| 1 | train1 | plma1 | plmasvg1 | test1 |
| 2 | train2 | plma2 | plmasvg2 | test2 |
| 3 | train3 | plma3 | plmasvg3 | test3 |
| 4 | train4 | plma4 | plmasvg4 | test4 |
| 5 | train5 | plma5 | plmasvg5 | test5 |
| 6 | train6 | plma6 | plmasvg6 | test6 |
| 7 | train7 | plma7 | plmasvg7 | test7 |
| 8 | train8 | plma8 | plmasvg8 | test8 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Grammar files obtained with different criteria
| split \ k,l | 1,1 | 2,2 | 3,3 | 4,4 | 5,5 | 6,6 | 7,7 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | |
| 0 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 1 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 2 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 3 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 4 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 5 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 6 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 7 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 8 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
Protein Family : M-Phase inducer
Cross-validation files
| split | training sequences | plma file | plma svg | test sequences |
|---|---|---|---|---|
| 0 | train0 | plma0 | plmasvg0 | test0 |
| 1 | train1 | plma1 | plmasvg1 | test1 |
| 2 | train2 | plma2 | plmasvg2 | test2 |
| 3 | train3 | plma3 | plmasvg3 | test3 |
| 4 | train4 | plma4 | plmasvg4 | test4 |
| 5 | train5 | plma5 | plmasvg5 | test5 |
| 6 | train6 | plma6 | plmasvg6 | test6 |
| 7 | train7 | plma7 | plmasvg7 | test7 |
| 8 | train8 | plma8 | plmasvg8 | test8 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Grammar files obtained with different criteria
| split \ k,l | 1,1 | 2,2 | 3,3 | 4,4 | 5,5 | 6,6 | 7,7 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | |
| 0 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 1 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 2 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 3 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 4 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 5 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 6 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 7 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 8 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
Protein Family : Zinc finger
Cross-validation files
| split | training sequences | plma file | plma svg | test sequences |
|---|---|---|---|---|
| 0 | train0 | plma0 | plmasvg0 | test0 |
| 1 | train1 | plma1 | plmasvg1 | test1 |
| 2 | train2 | plma2 | plmasvg2 | test2 |
| 3 | train3 | plma3 | plmasvg3 | test3 |
| 4 | train4 | plma4 | plmasvg4 | test4 |
| 5 | train5 | plma5 | plmasvg5 | test5 |
| 6 | train6 | plma6 | plmasvg6 | test6 |
| 7 | train7 | plma7 | plmasvg7 | test7 |
| 8 | train8 | plma8 | plmasvg8 | test8 |
| 9 | train9 | plma9 | plmasvg9 | test9 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Grammar files obtained with different criteria
| split \ k,l | 1,1 | 2,2 | 3,3 | 4,4 | 5,5 | 6,6 | 7,7 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | LS | LCS | |
| 0 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 1 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 2 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 3 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 4 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 5 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 6 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 7 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 8 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
| 9 | g | g | g | g | g | g | g | g | g | g | g | g | g | g |
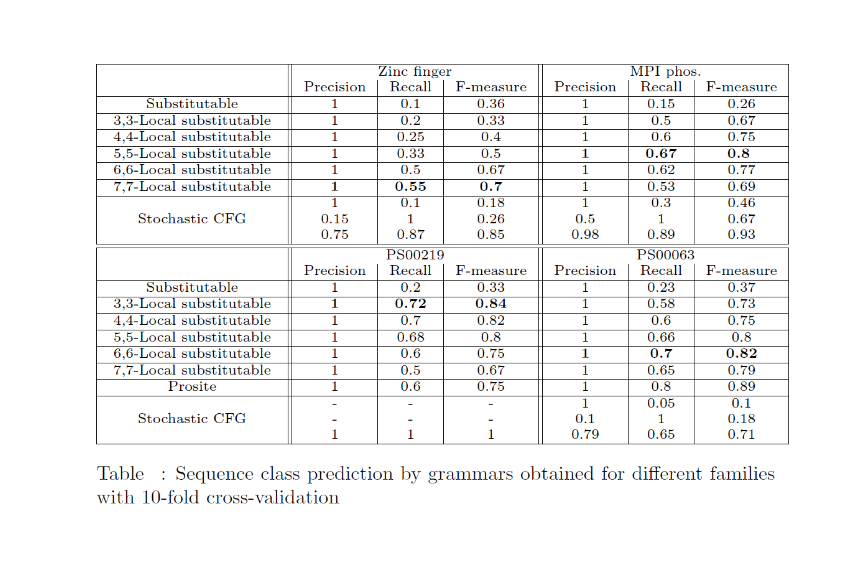
Results
![]()
The programs are available for academic usage upon request, please send us a mail : gaelle[dot]garet[at]inria[dot]fr