

PPalign:
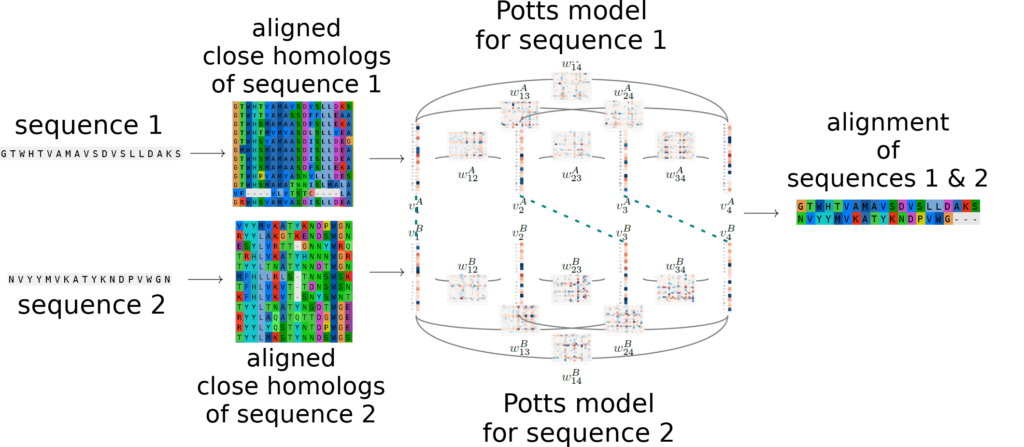
PPalign [1] performs Potts to Potts alignments of proteins taking into account residues coevolution. It relies on the efficient Integer Linear Programming solver originally designed by Wohlers, Andonov, Klau, Malod-Dognin and Yanev for protein structure alignment [2,3] to provide sequence alignments and similarity scores in tractable time.
GitHub repository
PPalign is available here: https://github.com/htalibart/ppsuite
Supplementary data
The benchmark for paper [1] is available here.
Each Potts models was built using the following command (replacing sequence.fasta with the path of the sequence file and uniclust30_2018_08 with the path of UniClust30 database):
makepotts -s sequence.fasta -fetch -d uniclust30_2018_08 --trimal_gt 0.5 --insert_null_at_trimmed -f output_folder
Bibliography
[1] Talibart, H, & Coste, F. PPalign: optimal alignment of Potts models representing proteins with direct coupling information, BMC Bioinformatics (2021).
[2] Wohlers, I., Andonov, R., & Klau, G. W. (2012). DALIX: optimal DALI protein structure alignment. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 10(1), 26-36.
[3] Andonov, R., Malod-Dognin, N., & Yanev, N. (2011). Maximum contact map overlap revisited. Journal of Computational Biology, 18(1), 27-41.